

Editor’s Note:At the recently concluded 2026 Annual Meeting of the American Association for Cancer Research (AACR), Qilu Pharmaceutical presented 12 innovative oncology programs, including one oral presentation and eleven poster presentations. These studies span a wide range of tumor types, including gynecologic cancers, breast cancer, esophageal cancer, lung cancer, head and neck cancers, colorectal cancer, prostate cancer, diffuse large B-cell lymphoma (DLBCL), and acute myeloid leukemia (AML).The portfolio covers multiple advanced therapeutic modalities, including mono- and bispecific antibody–drug conjugates (ADCs), trispecific T-cell engagers (TCEs), natural killer cell engagers (NKCEs), and novel small-molecule inhibitors. Targeting more than ten key molecules—such as CLDN6, 5T4, B7-H3, PARG, and KIF18A—these programs collectively highlight the depth of innovation and cutting-edge progress of Chinese research in the field of cancer therapeutics.
Message
(Video message)
Dr. Weikang Tao (Executive Vice President of Qilu Pharmaceutical Group and Head of Global Innovative R&D):
“At this year’s AACR meeting, Qilu Pharmaceutical presented the R&D progress of 12 innovative drug programs, including an oral presentation on a novel ADC. After several years of dedicated effort, we have made significant advances across small-molecule targeted therapies, ADCs, and T-cell engagers. Encouragingly, some of these candidates have demonstrated favorable tolerability, safety, and efficacy in clinical studies.
The programs showcased here represent only part of our pipeline, and more innovative therapies will enter clinical development in the future. We hope these medicines will address unmet clinical needs and ultimately benefit patients. We also look forward to collaborating with clinical experts, basic researchers, and industry peers to further advance China’s innovative drug development and bring these therapies to the global stage.”

△Dr. Weikang Tao’s Team at AACR
Monospecific and Bispecific ADCs: Differentiated Target Design Driving Efficacy Breakthroughs
1. CLDN6-Targeting ADC QLS5132 Demonstrates Promising Preliminary Efficacy in PROC

△Professor Tao Zhu from Zhejiang Cancer Hospital Presented QLS5132 at AACR
QLS5132 is a first-in-class ADC designed to combine a CLDN6-targeting monoclonal antibody with a topoisomerase I inhibitor (Top1i) payload, enabling dual mechanisms of action—precise targeting and potent cytotoxicity—against tumor cells with high CLDN6 expression. Unlike other ADCs in development targeting the same antigen that employ MMAE or PBD payloads, the differentiated design of QLS5132 achieves a more favorable balance between efficacy and safety.
Importantly, QLS5132 does not rely on CLDN6 expression levels for patient selection in clinical settings. Patients may benefit from treatment regardless of whether their tumors exhibit high or low CLDN6 expression.
Data presented at AACR demonstrated encouraging preliminary results from a Phase I clinical trial in patients with advanced platinum-resistant ovarian cancer (PROC). Among 18 evaluable patients, the objective response rate (ORR) reached 50.0%, and the disease control rate (DCR) was 94.4%. At the recommended Phase II dose (RP2D), efficacy was further improved (ORR 55.6%, DCR 100%), with a manageable safety profile. The most common treatment-related adverse events (TRAEs) were grade 1–2, with no grade 4–5 events observed, and no cases of interstitial lung disease (ILD) or other notable toxicities reported.
Additionally, preliminary clinical findings showed no significant correlation between treatment response and CLDN6 expression levels, suggesting that biomarker testing may not be required in the future. This could potentially broaden patient access and enable more individuals to benefit from the therapy.
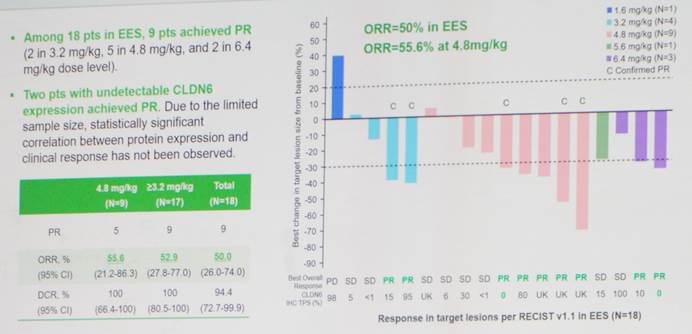
△Best Overall Response to QLS5132 in PROC
2. 5T4-Targeting ADC LUA004 Offers a Broader Therapeutic Window for Squamous Cell Carcinoma

△LUA004 Poster Presentation
LUA004 is a novel 5T4-targeting ADC with potential for pan-tumor applications. Its target, 5T4, is highly expressed across multiple squamous cell carcinomas, including esophageal squamous cell carcinoma (ESCC), head and neck squamous cell carcinoma (HNSCC), and lung squamous cell carcinoma (LUSC).
LUA004 incorporates a proprietary, novel microtubule inhibitor (MTI) payload. While maintaining strong antitumor efficacy, this payload significantly reduces the risks of hepatotoxicity and myelosuppression and demonstrates faster systemic clearance compared with MMAE. In addition, the ADC is engineered with an antibody optimized for enhanced binding affinity and internalization.
Through the synergistic optimization of target selection, payload design, and antibody engineering, LUA004 achieves a broader therapeutic window than conventional MMAE-based ADCs. In cynomolgus monkey toxicity studies, the highest non-severely toxic dose (HNSTD) reached ≥20 mg/kg administered Q3W×4, compared with <10 mg/kg for vedotin-based ADCs given as a single dose, highlighting its markedly improved tolerability and expanded therapeutic window.
Furthermore, LUA004 demonstrated robust antitumor activity across multiple 5T4-expressing models, including cell line–derived xenograft (CDX) and patient-derived xenograft (PDX) models of ESCC, HNSCC, and LUSC. These findings provide strong support for its ongoing Phase I clinical trial (NCT07467629).
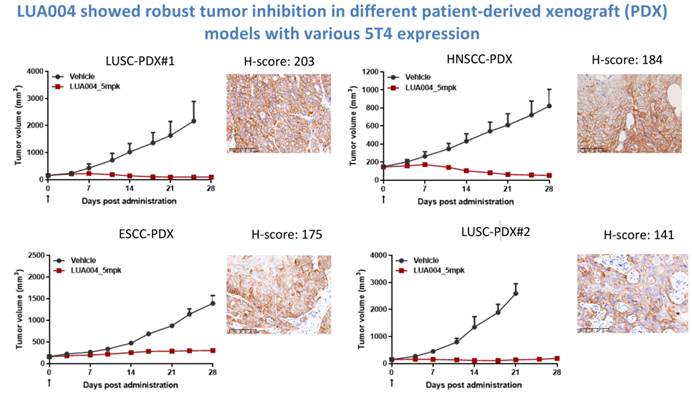
△LUA004 Demonstrates Antitumor Activity in PDX Models
3. EGFR/c-MET Bispecific ADC LUA005 Shows Antitumor Activity and a Broader Therapeutic Window in Solid Tumors
△LUA005 Poster Presentation
LUA005 is an EGFR/c-MET bispecific ADC featuring a unique bivalent, dual-arm design. It exhibits low affinity but high avidity to EGFR, coupled with high affinity to c-MET. This design helps reduce on-target off-tumor toxicity while maintaining potent cytotoxic activity against tumors with EGFR/c-MET co-expression or those exhibiting low EGFR expression.
The cytotoxic payload is a clinically validated, proprietary Top1i, characterized by high stability and rapid systemic clearance, further contributing to an improved safety profile.
Data presented at this meeting showed that, compared with a benchmark molecule (BMK) targeting the same antigens, LUA005 demonstrated superior safety in cynomolgus monkeys, with a tolerated single dose of ≥60 mg/kg versus <30 mg/kg for BMK. In addition, LUA005 exhibited strong antitumor activity across multiple pretreated and resistant CDX and PDX models, including non-small cell lung cancer (NSCLC), ESCC, HNSCC, and colorectal cancer.
LUA005 has now advanced into a Phase I clinical trial (CRT20260037).
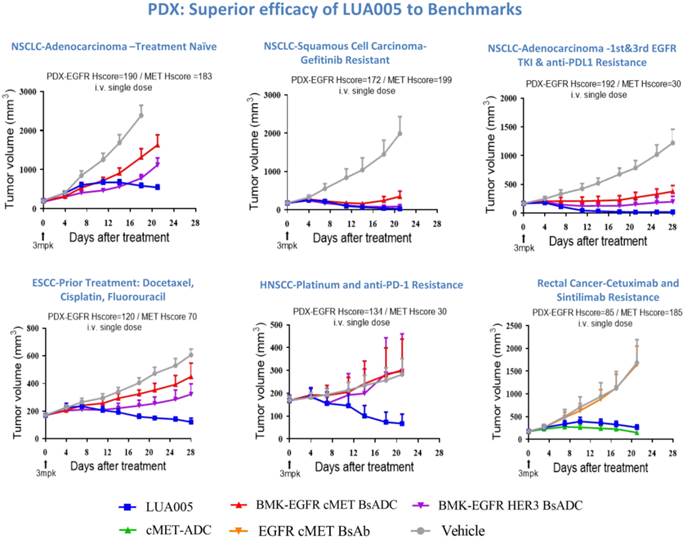
△LUA005 Demonstrates Antitumor Activity in PDX Models
4. LUA006,A Bispecific EGFR/B7-H3 ADC with Dual-Payload to Overcome Resistance and Tumor Heterogeneity
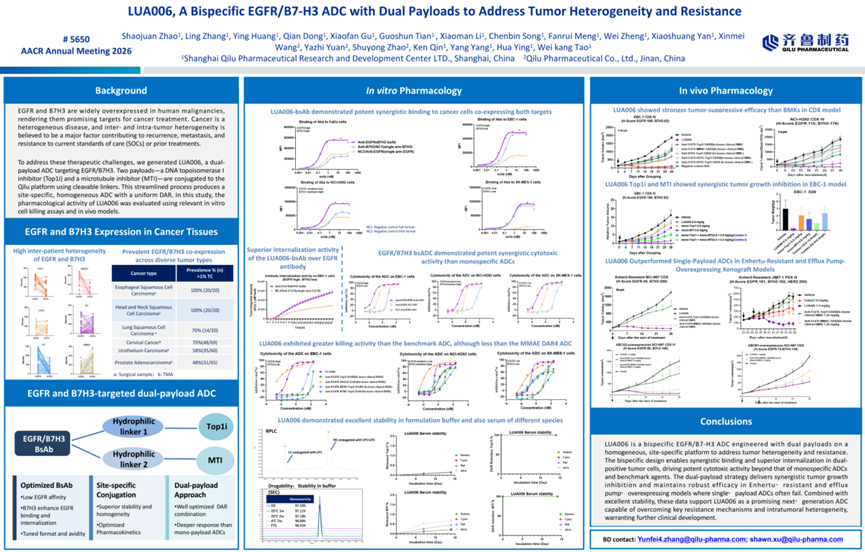
△LUA006 Poster Presentation
LUA006 is a first-in-class bispecific ADC targeting EGFR and B7-H3, which bears two distinct payloads—— a Top1i and a MTI. Its bispecific antibody (bsAb) design enables cooperative binding and enhanced internalization, while maintaining a uniform drug-to-antibody ratio (DAR).
In vitro studies demonstrated that the bsAb component of LUA006 exhibits significantly stronger binding across cancer cell lines co-expressing EGFR and B7-H3 at varying expression levels compared with monospecific controls, along with superior internalization efficiency, thereby helping to address tumor heterogeneity.
The dual-payload design produces a synergistic cytotoxic effect, substantially exceeding that of monospecific ADCs and clinical benchmark agents, while maintaining strong molecular stability. In vivo, LUA006 achieved greater tumor growth inhibition than clinical-stage comparator drugs (BMKs) in both CDX and PDX models. Notably, in models resistant to Enhertu and in those with overexpression of efflux pumps, LUA006 demonstrated more effective tumor suppression than single-payload ADCs, highlighting its ability to overcome resistance mechanisms.
Overall, the dual-target, dual-payload strategy of LUA006 effectively addresses tumor heterogeneity and drug resistance, while maintaining excellent stability, positioning it as a highly promising next-generation ADC candidate.
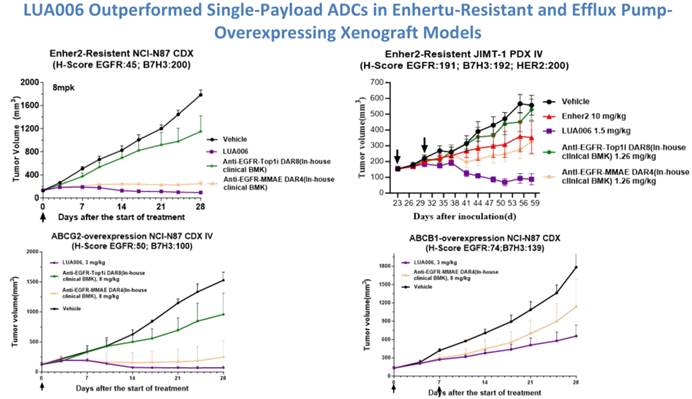
△LUA006 Demonstrates Superior Antitumor Activity Compared with Single-Payload ADCs in Enhertu-Resistant and Efflux Pump–Overexpressing Models
5. CDH17/c-MET Bispecific ADC LUA011 Enhances Therapeutic Efficacy in Gastrointestinal Tumors
△LUA011 Poster Presentation
LUA011 is a first-in-class bispecific ADC targeting CDH17 and c-MET. The CDH17-binding arm is designed with monovalent affinity to reduce excessive binding in the gastrointestinal tract, thereby minimizing on-target, off-tumor toxicity. In contrast, the c-MET arm is optimized for enhanced internalization, enabling more efficient cellular uptake of the drug.
The cytotoxic payload is a Top1i, which is less susceptible to efflux pump–mediated resistance and may therefore help overcome certain resistance mechanisms. As CDH17 and c-MET are frequently co-expressed in gastrointestinal tumors but rarely co-expressed in normal tissues, this dual-targeting strategy is expected to improve efficacy while reducing toxicity.
In cynomolgus monkeys, LUA011 demonstrated superior tolerability compared with benchmark agents (BMK, a c-MET ADC, and SOT109, a CDH17 ADC), with a maximum tolerated dose (MTD) of ≥30 mg/kg. Moreover, in colorectal cancer models with low c-MET expression, LUA011 was able to overcome the limitations of single-target c-MET ADCs and exhibited enhanced antitumor activity.
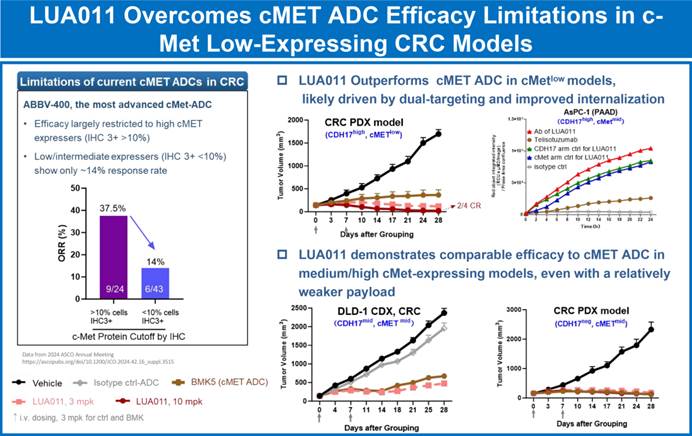
△LUA011 Demonstrates Strong Activity in Colorectal Cancer Models with Low c-MET Expression
Innovative Engager Architectures Targeting Both Solid and Hematologic Malignancies
1. PSMA/STEAP1/CD3 Tri-specific TCE QLS2401 Offers a Potential New Approach for the Treatment of mCRPC
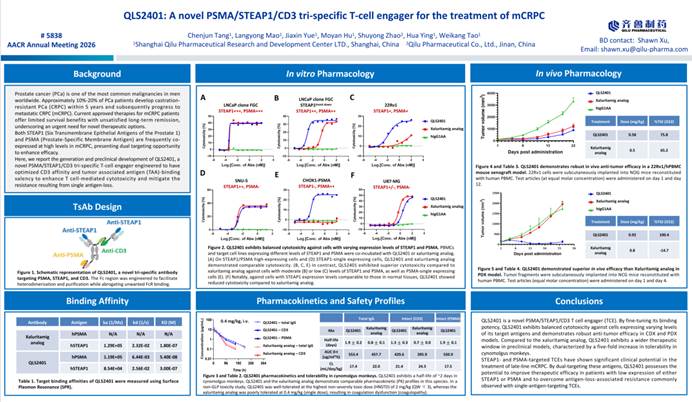
△QLS2401 Poster Presentation
QLS2401 is a PSMA/STEAP1/CD3 tri-specific TCE. By simultaneously targeting PSMA and STEAP1—two antigens frequently co-expressed in metastatic castration-resistant prostate cancer (mCRPC)—it is designed to overcome resistance driven by single-antigen loss. The CD3-binding domain has been affinity-tuned to reduce the risk of excessive T-cell activation, while the optimized valency for tumor-associated antigen (TAA) binding enhances cytotoxic activity against tumors with low antigen density.
Preclinical data show that, compared with a Xaluritamig-like comparator, QLS2401 demonstrates comparable antitumor activity in cell lines with high PSMA/STEAP1 expression, and greater cytotoxicity in models with medium to low expression. In cynomolgus monkeys, QLS2401 exhibited a wider therapeutic window (HNSTD of 2 mg/kg, QW×3), whereas the comparator induced coagulation abnormalities at a single dose of 0.4 mg/kg.
In both mouse CDX and PDX tumor models, QLS2401 also demonstrated superior antitumor efficacy compared with the benchmark agent.
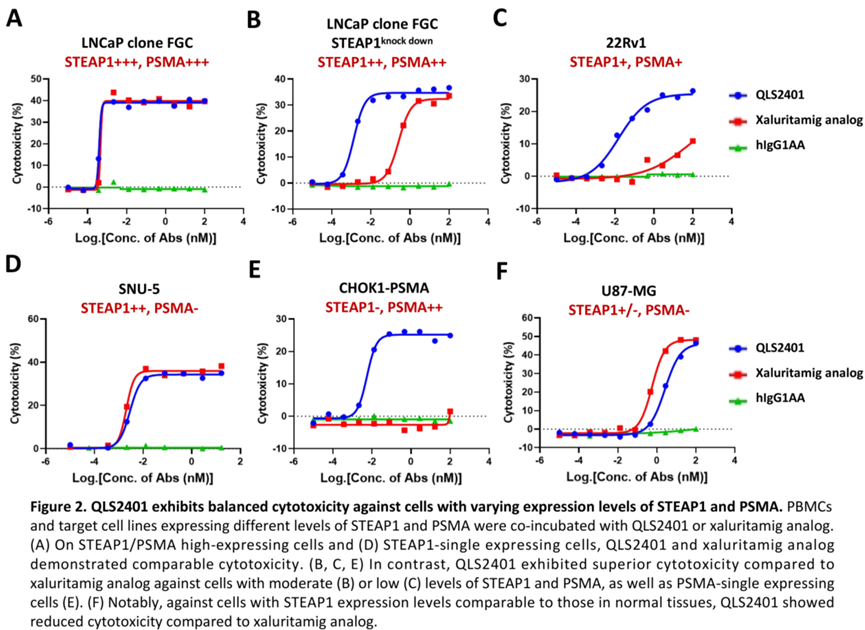
△QLS2401 Demonstrates Consistent Cytotoxic Activity Across Cell Lines with Varying Levels of STEAP1 and PSMA Expression
2. PSMA/CD3/CD2 Tri-specific TCE QL535: First-in-Class CD2 Targeting with Enhanced Efficacy and Reduced CRS Risk
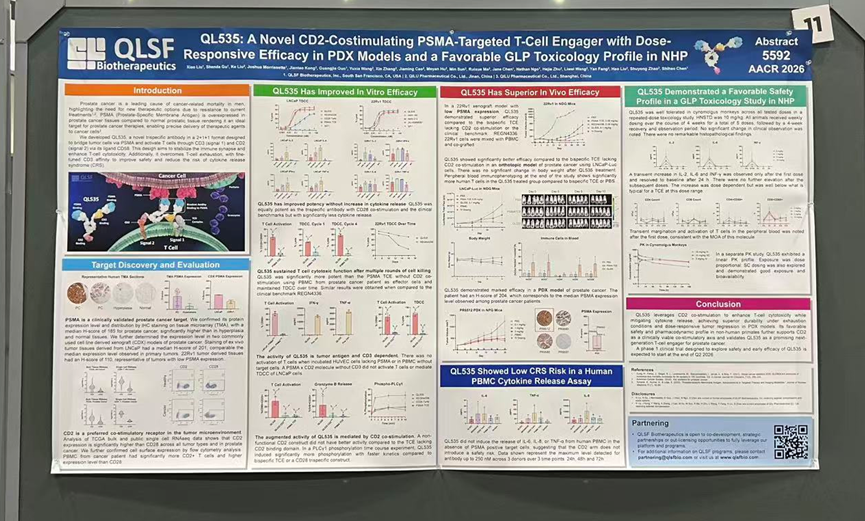
△QL535 Poster Presentation
QL535 is a novel tri-specific antibody with a 2+1+1 format architecture. In addition to targeting tumor cells via PSMA and activating T cells through CD3, it innovatively incorporates CD2-mediated co-stimulatory signaling. This design enhances the stability of T cell–tumor cell interactions, augments T-cell cytotoxic function, and helps overcome T-cell exhaustion. At the same time, fine-tuning of CD3 affinity reduces the risk of cytokine release syndrome (CRS).
These advantages are supported by preclinical data, which show that QL535 outperforms CD2-negative bispecific antibodies and clinical benchmark agents in enhancing T-cell–mediated cytotoxicity and reversing T-cell exhaustion.
In GLP toxicology studies in non-human primates, QL535 was well tolerated at all tested dose levels, with only transient and mild cytokine elevations observed following the first administration, supporting its favorable safety profile.
Overall, by integrating CD2 co-stimulation, QL535 achieves a strong balance between efficacy and safety. A Phase I clinical trial is planned to initiate in the second quarter of 2026 to further evaluate its early efficacy and safety.
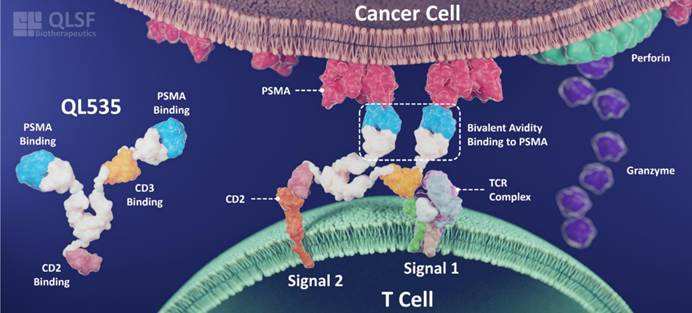
△Mechanism of Action of QL535
3. CD79b/CD20/CD3 Tri-specific TCE QLS2313 Shows Potential to Overcome CD20 Resistance in DLBCL
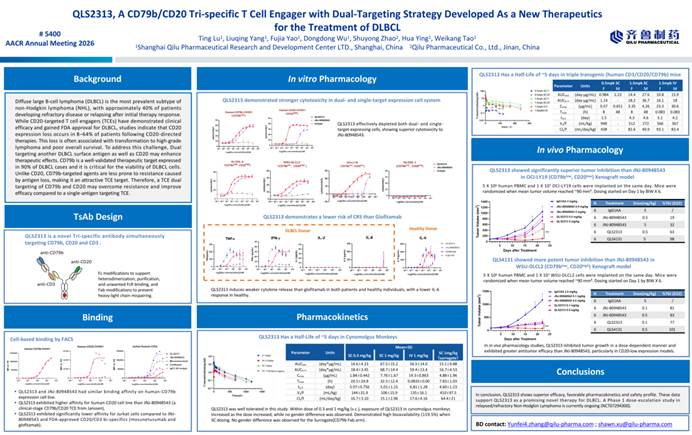
△QLS2313 Poster Presentation
To address antigen-loss–mediated resistance following CD20-targeted therapy in diffuse large B-cell lymphoma (DLBCL), QLS2313 is a tri-specific TCE targeting CD79b, CD20, and CD3. The inclusion of CD79b is designed to overcome resistance and synergize with CD20 for coordinated tumor killing.
Compared with JNJ-80948543, which is currently under clinical investigation, QLS2313 exhibits higher affinity for CD20 and lower affinity for CD3, translating into stronger cytotoxic activity in vitro. Relative to the approved agent Glofitamab, QLS2313 induces lower levels of inflammatory cytokines such as IL-6, suggesting a reduced risk of cytokine release syndrome (CRS).
In humanized mouse xenograft models of DLBCL with CD79blow/CD20low or CD79blow/CD20high expression, QLS2313 demonstrated superior efficacy compared with JNJ-80948543.
A Phase I dose-escalation study in relapsed/refractory non-Hodgkin lymphoma (NHL) (NCT07294300) is currently ongoing, with promising potential.
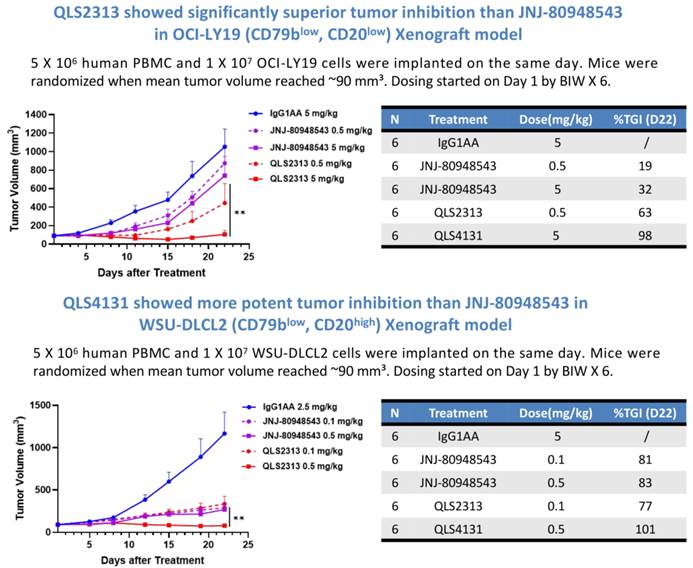
△QLS2313 Demonstrates Antitumor Efficacy in DLBCL Models
4. CD70/NKp46 Bispecific NK Cell Engager (NKCE) QLS2309 Offers a Potential New Option for Hematologic Malignancies
△QLS2309 Poster Presentation
CD70 is a well-established therapeutic target in acute myeloid leukemia (AML). However, approximately one-third of AML patients exhibit high expression of CD64, which has high affinity for afucosylated Fc regions and can impair antibody-dependent cell-mediated cytotoxicity (ADCC) of conventional monoclonal antibodies.
QLS2309 is a CD70/NKp46 bispecific NK cell engager (NKCE) that functionally targets three molecules—CD70, NKp46, and CD16a. It incorporates a wild-type Fc domain, enabling binding to activating receptors CD16a and NKp46 on NK cells, thereby activating NK cells while bypassing CD64 engagement.
Preclinical studies demonstrated that QLS2309 induces stronger ADCC activity than the ADCC-enhanced monoclonal antibody Cusatuzumab across AML cell lines with both CD70high/CD64+ and CD70low/CD64– expression profiles. These findings were further confirmed in xenograft models of AML and lymphoma, where QLS2309 consistently showed superior antitumor efficacy.
A Phase I dose-escalation study of QLS2309 in patients with CD70-positive relapsed/refractory hematologic malignancies (NCT07173595) is currently underway.
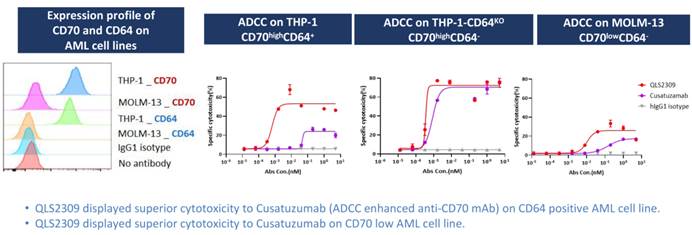
△Comparison of ADCC Activity Between QLS2309 and Cusatuzumab Across Different Cell Lines
Novel Small-Molecule Inhibitors: Precisely Targeting Tumor-Specific Vulnerabilities
1. KIF18A Inhibitor QLS1303 Demonstrates Potent Antitumor Activity in CIN+ Tumor Models
△QLS1303 Poster Presentation
KIF18A is a protein essential for mitosis in chromosomal instability (CIN) positive tumor cells but is dispensable in normal diploid cells, creating a therapeutic opportunity based on synthetic lethality. QLS1303 is a potent, selective, and orally bioavailable KIF18A inhibitor. In vitro, it selectively inhibits the proliferation of CIN+ tumor cells while having minimal impact on normal cells.
Across multiple tumor models—including platinum-sensitive ovarian cancer with TP53 mutation and CCNE1 amplification, microsatellite-stable (MSS) colorectal cancer with TP53 mutation, and platinum-resistant ovarian cancer with TP53 mutation and BRCA silencing—QLS1303 demonstrated superior efficacy compared with the benchmark compound AMG 650.
From a safety perspective, QLS1303 showed no significant inhibition across 46 off-targets (IC₅₀ > 10 μM), indicating a wide therapeutic window. These findings support its potential as a preferred therapeutic option for CIN+ tumors.
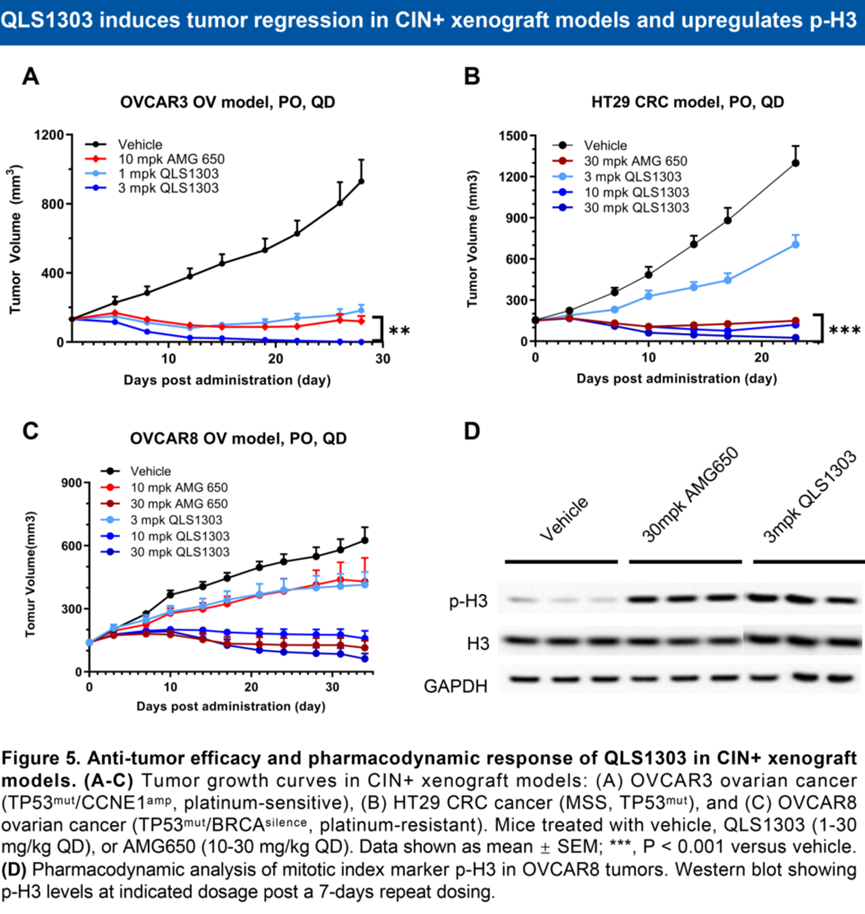
△QLS1303 Demonstrates Antitumor Activity in CIN+ Models
2. PARG Inhibitor QLS1403 Shows Potential to Overcome Resistance to PARP Inhibitors and T-DXd
△QLS1403 Poster Presentation
PARG is a key enzyme in the DNA damage repair pathway, responsible for hydrolyzing the poly(ADP-ribose) (PAR) chains generated by PARP during DNA damage. This process facilitates the dissociation of DNA repair complexes and enables subsequent repair steps.
In cells with homologous recombination deficiency (HRD), the selective PARG inhibitor QLS1403 induces accumulation of PAR chains, leading to replication fork stalling and failure of DNA repair, ultimately triggering cell death.
QLS1403 demonstrates approximately 10-fold greater inhibitory activity against PARG compared with the clinical-stage comparator IDE-161. In cell lines with acquired resistance to Olaparib or T-DXd, QLS1403 exhibited potent antiproliferative activity.
In CDX models of HRD-positive breast and ovarian cancers with primary resistance to PARP inhibitors (PARPi), QLS1403 induced tumor regression at lower doses than IDE-161. This effect was accompanied by dose-dependent accumulation of intratumoral PAR, highlighting its potential to overcome resistance to both PARP inhibitors and ADC therapies.
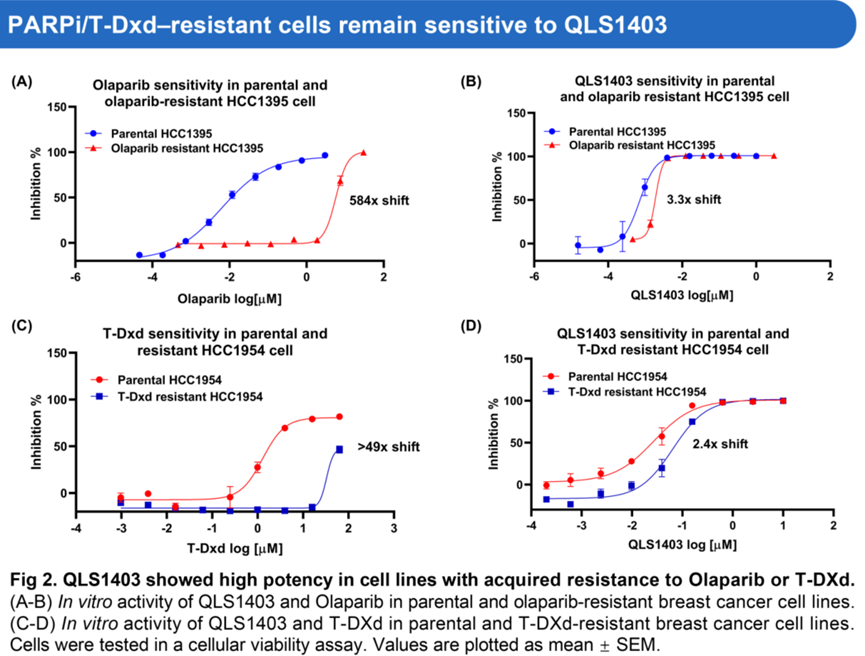
△QLS1403 Demonstrates the Ability to Overcome Resistance to PARP Inhibitors and T-DXd
3. PI3Kα Inhibitor QLS1522 Offers a Potential New Option for PIK3CA-Mutant Breast Cancer
△QLS1522 Poster Presentation
QLS1522 is a potent, mutant-selective allosteric inhibitor of PI3Kα that preferentially targets PIK3CA mutations while sparing wild-type PI3Kα, thereby significantly widening the therapeutic window without compromising antitumor efficacy.
In vitro, QLS1522 exhibits nanomolar inhibitory activity against PIK3CA mutations (e.g., H1047R), with substantially lower activity against wild-type PI3Kα and other isoforms (β, γ, δ). Its high specificity was further confirmed in a kinase selectivity panel covering 437 kinases.
In preclinical in vivo studies, QLS1522 demonstrated robust antitumor activity across multiple ER-positive breast cancer CDX models harboring PIK3CA mutations, including both kinase-domain and helical-domain alterations, without causing significant hyperglycemia in mice.
When combined with CDK4/6 inhibitors and endocrine therapy, QLS1522 showed synergistic tumor growth inhibition in PIK3CAH1047R ER+/HER2− breast cancer models. Notably, the combination of QLS1522 with a CDK4/6 inhibitor and Fulvestrant achieved the greatest tumor suppression, significantly outperforming the combination of a CDK4/6 inhibitor plus fulvestrant alone.
From a safety perspective, QLS1522 did not induce hyperglycemia at therapeutically effective doses in mice and demonstrated favorable pharmacokinetic (PK) properties across mice, rats, and dogs, indicating a broad therapeutic window. The program has now advanced to the IND-enabling stage.
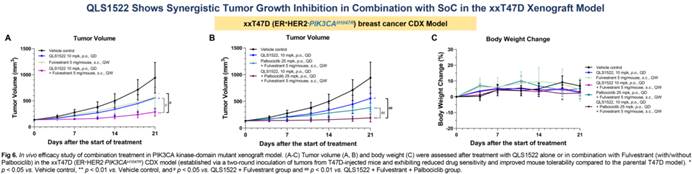
△QLS1522 Combination Strategies Show Greater Development Potential
Conclusion
The presentation of these 12 research programs at the 2026 AACR Annual Meeting highlights the comprehensive R&D capabilities of Chinese biopharmaceutical companies, spanning the full spectrum from preclinical discovery to clinical development.
In the ADC field, Chinese researchers are advancing differentiated designs aimed at improving therapeutic index. In T-cell and NK-cell engager therapies, multi-target and co-stimulatory strategies are being leveraged to enhance efficacy, overcome resistance, and better manage toxicity. In small-molecule drug discovery, efforts are focused on novel targets and innovative molecular designs.
Together, these advancements are driving China’s transition from a “fast follower” to a true innovator capable of delivering first-in-class and best-in-class therapies. They also open new avenues for optimizing treatment strategies for cancer patients worldwide.
China’s innovative drug development landscape is steadily progressing toward the global forefront, and further clinical validation in the years ahead is highly anticipated.

