
Editor’s Note: Primary Biliary Cholangitis (PBC) is a chronic cholestatic liver disease mediated by autoimmunity, with its pathogenesis not yet fully understood. Traditionally, PBC was thought to predominantly affect Western populations, but in recent decades, there has been an increasing number of reported cases and published literature in the Asia-Pacific region, escalating the disease burden. In response, the Asia-Pacific Association for the Study of the Liver (APASL) released clinical guidelines for PBC last year to better guide clinical practice. Recently, at the 32nd APASL Annual Conference, Professor Jia Jidong from the Capital Medical University Affiliated Beijing Friendship Hospital, China, delivered an insightful academic lecture titled “Latest Advances in Primary Biliary Cholangitis”.
PBC is a chronic progressive autoimmune cholestatic disease, primarily targeting the liver. Its main pathological change is non-suppurative inflammation in the intrahepatic small bile ducts, eventually leading to liver fibrosis and cirrhosis. In recent years, as understanding of the disease deepens, along with the development and widespread availability of autoantibody testing techniques, the overall incidence of PBC has been rising annually. It is estimated that nearly 100,000 people are diagnosed with PBC worldwide each year [1]. Currently, China lacks population-based epidemiological data on PBC, but the estimated prevalence in China is about 20.5 per 100,000, ranking second in the Asia-Pacific region, just behind Japan [2-3].
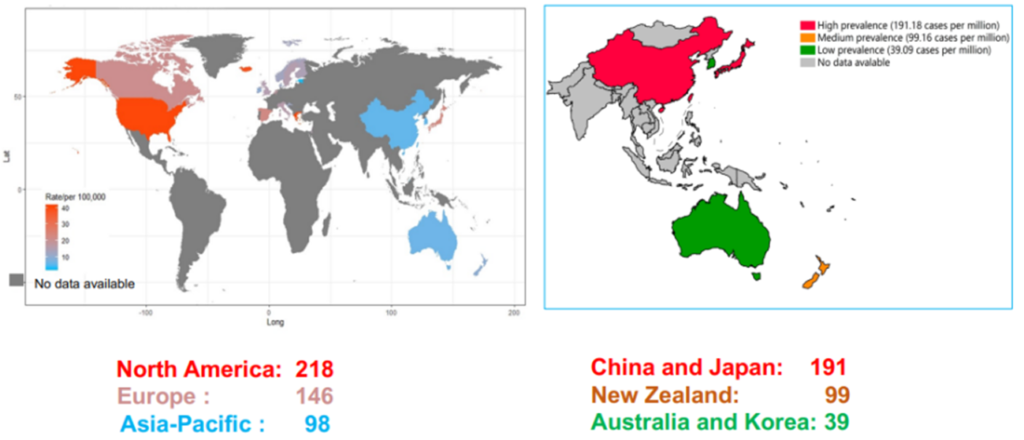
Figure 1. Global Prevalence of PBC
(Sourced from the speaker’s presentation)
Diagnosis
PBC is a global disease, primarily affecting middle-aged and older women. Clinical symptoms of PBC patients are highly heterogeneous, mainly manifesting as itching, fatigue, jaundice, etc., with severe cases leading to liver decompensation. Many patients in the early stages of PBC have no obvious clinical symptoms, making the disease’s clinical presentation elusive. Some patients are already in a stage of liver cirrhosis at the time of discovery.
The etiology and pathogenesis of PBC have not been fully elucidated yet. They are possibly related to immune dysregulation caused by genetic factors and their interaction with environmental factors. Its clinical characteristics include high titers of anti-mitochondrial antibodies (AMA) in serum, elevated bile enzymes, and characteristic liver pathology; immunological features include elevated serum alkaline phosphatase (ALP) and gamma-glutamyl transpeptidase (GGT), increased immunoglobulin M (IgM), and the histological feature of non-suppurative destructive cholangitis of interlobular bile ducts [4]. In particular, the presence of AMA or AMA-M2 is highly valuable for diagnosing PBC in clinical populations, with both sensitivity and specificity exceeding 90%.
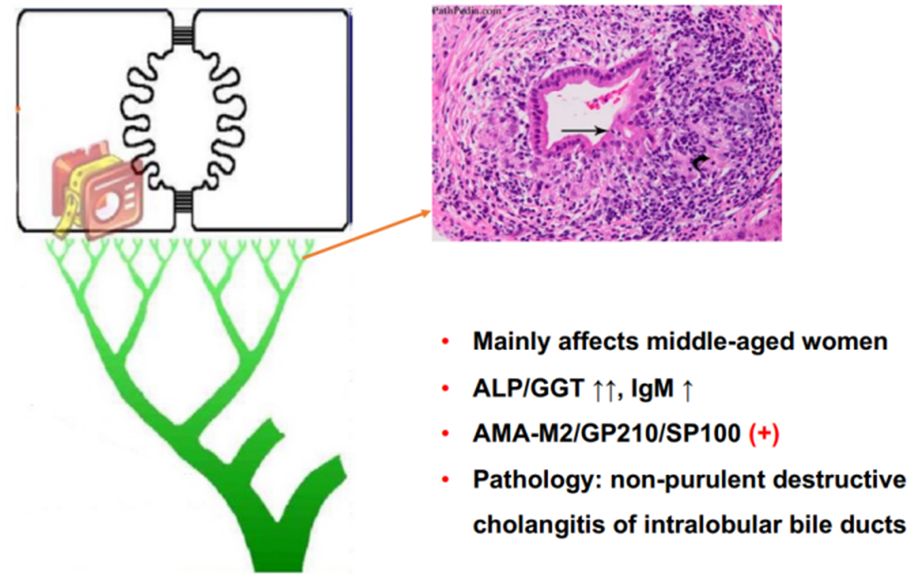
Figure 2. Disease Characteristics of PBC
(Sourced from the speaker’s presentation)
In summary, the diagnosis of Primary Biliary Cholangitis (PBC) relies on a comprehensive assessment of biochemical, immunological, radiological, and histological examinations. The 2022 edition of the Asia-Pacific Association for the Study of the Liver (APASL) Clinical Guidelines for PBC recommends that a diagnosis of PBC can be confirmed when at least two of the following three criteria are met: (1) Elevated biochemical indicators of cholestasis (mainly ALP and GGT) and radiological exclusion of intrahepatic and extrahepatic biliary obstruction; (2) Positive for anti-mitochondrial antibodies (AMA), or other PBC-specific anti-nuclear antibodies (ANA), such as anti-sp100 or anti-gp210 antibodies; (3) Liver biopsy showing non-suppurative destructive cholangitis primarily involving interlobular bile ducts.
It is important to note that serum AMA is a specific marker for the diagnosis of PBC, but PBC cannot be diagnosed solely based on AMA. Patients who are AMA-positive but have normal liver biochemical indicators should be closely followed up, with annual re-evaluations of serological markers. Liver biopsy may be necessary for definitive diagnosis of the disease.
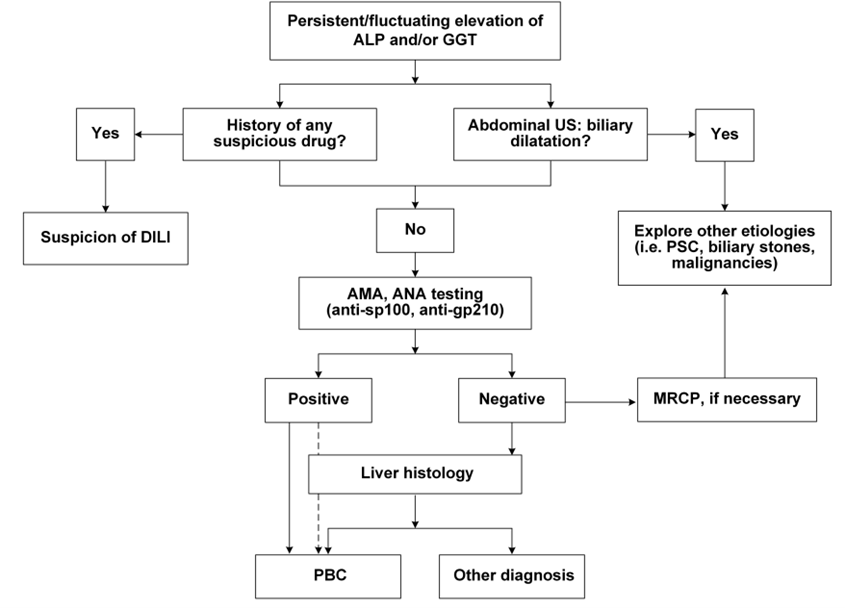
Figure 3. Clinical Diagnostic Pathway for PBC
(Sourced from the speaker’s presentation)
Treatment
Long-term treatment with Ursodeoxycholic Acid (UDCA) at a dosage of 13-15 mg/kg/day is the standard therapy for Primary Biliary Cholangitis (PBC). The mechanism of action of UDCA includes choleretic (bile-producing) effects, cellular protection, anti-inflammatory properties, and immune modulation. It is effective in improving biochemical markers in patients, alleviating pathological changes, and slowing the progression of the disease. More than 60% of patients show a favorable biochemical response after starting the medication. Research indicates that the use of UDCA prolongs transplant-free survival in PBC patients, regardless of their gender, age, disease stage, or response to treatment [5].
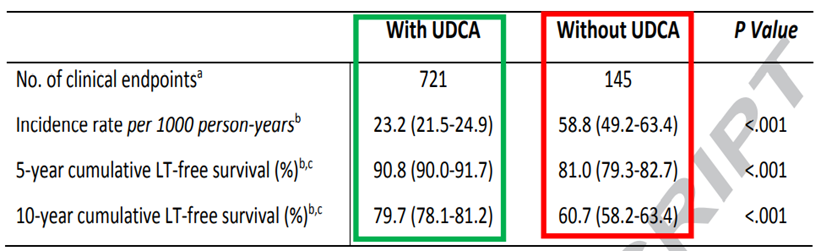
Figure 4. Clinical Treatment Efficacy of UDCA
(Sourced from the speaker’s presentation)
When using Ursodeoxycholic Acid (UDCA) for the treatment of Primary Biliary Cholangitis (PBC), the patient’s biochemical response should be evaluated after 6-12 months using common standards such as the Paris II criteria (suitable for early-stage PBC) and the Paris I criteria (suitable for late-stage PBC). If the patient meets these response criteria, there is typically a significant improvement in liver histology and long-term survival prognosis. Conversely, if there is a poor or no biochemical response after 6-12 months or longer of treatment, it may be necessary to consider additional second-line therapies such as Obeticholic Acid (OCA), fibrates, or budesonide.
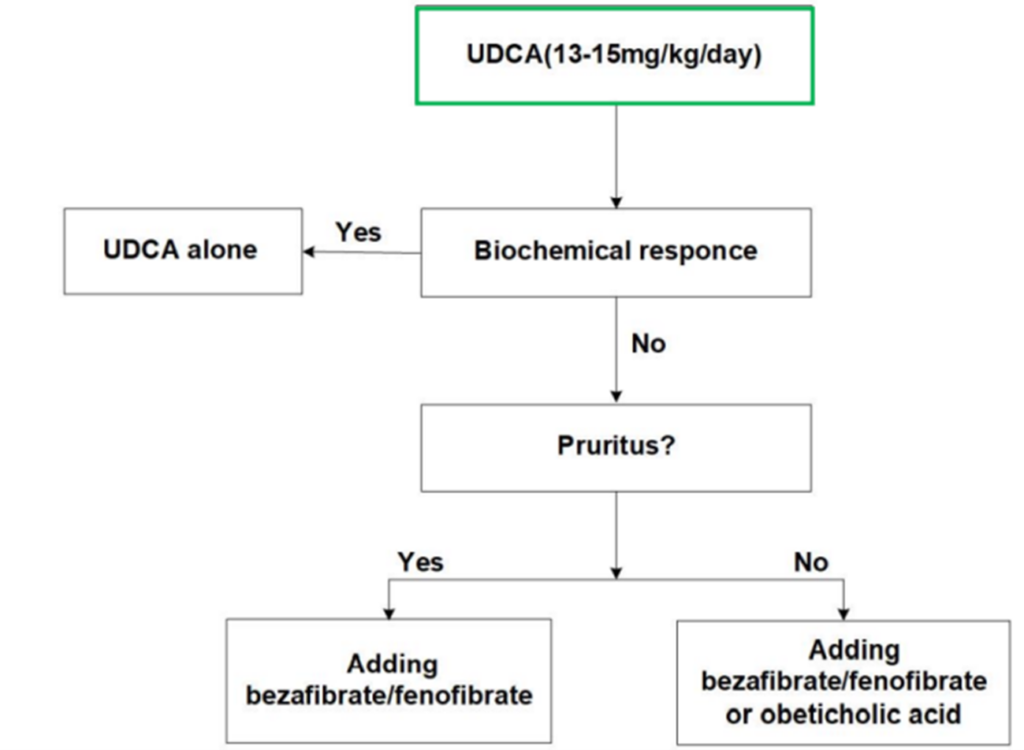
Figure 5. Clinical Treatment of PBC
(Sourced from the speaker’s presentation)
New Advances
Currently, the first-line treatment for Primary Biliary Cholangitis (PBC) is Ursodeoxycholic Acid (UDCA), but up to 40% of PBC patients show a poor response to UDCA treatment. Therefore, there is an urgent need to identify new drug targets and develop novel therapeutic agents. Bile acid accumulation plays a significant role in the development and progression of PBC. Effectively reducing bile stasis and decreasing bile acid toxicity are key directions for new drug development.
There are several key regulatory targets in the enterohepatic circulation of bile acids, such as Farnesoid X Receptor (FXR), Peroxisome Proliferator-Activated Receptors (PPARs), and the Ileal Bile Acid Transporter (IBAT). These targets can achieve therapeutic or improvement effects on PBC through different pathways and are currently considered the most promising drug targets.
FXR Agonists
Obeticholic Acid (OCA) is a semi-synthetic hydrophobic bile acid analog with high selectivity for FXR. It can inhibit the gene expression of the rate-limiting enzyme in bile acid synthesis, thereby regulating bile acid metabolism and reducing inflammation and liver fibrosis. Activated FXR inhibits the transcription of CYP7A1, thus regulating the synthesis, absorption, transport, and secretion of bile acids. Additionally, the activation of FXR promotes the production of Fibroblast Growth Factor 19 (FGF19), which acts on hepatocytes to inhibit the expression of CYP7A1, further suppressing bile acid production.
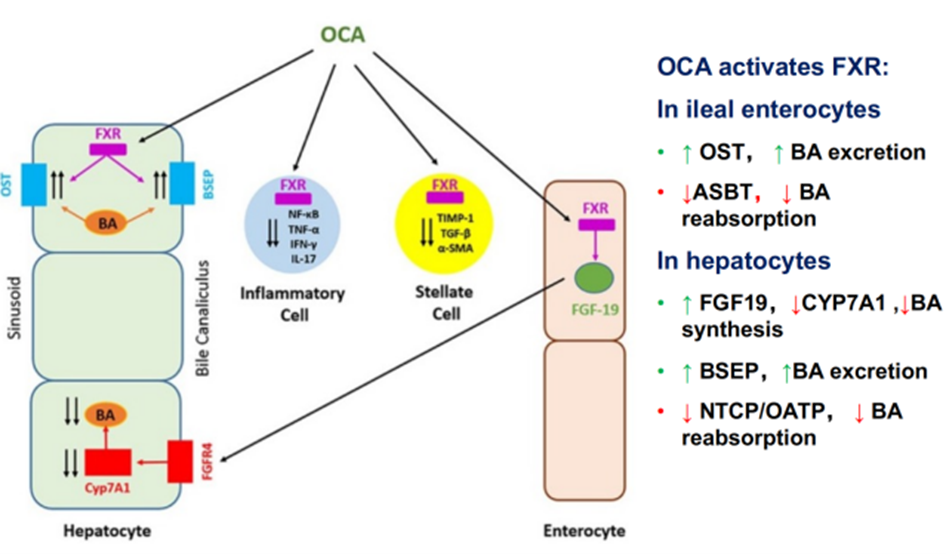
Figure 6. Mechanism of Action of Obeticholic Acid (OCA)
(Sourced from the speaker’s presentation)
A Phase III clinical trial showed [6] that adding Obeticholic Acid (OCA) significantly reduces serum ALP and TBIL levels in patients who respond poorly to or are intolerant of Ursodeoxycholic Acid (UDCA). Long-term follow-up results over three years also demonstrate the sustained efficacy of OCA (Figure 7). Apart from OCA, other novel FXR agonists, such as Tropifexor (LJN452), have shown promising results in animal studies and short-term clinical trials. These include their effectiveness in reducing bile stasis, improving liver biochemical markers, and inhibiting tissue inflammatory cell infiltration and fibrosis.
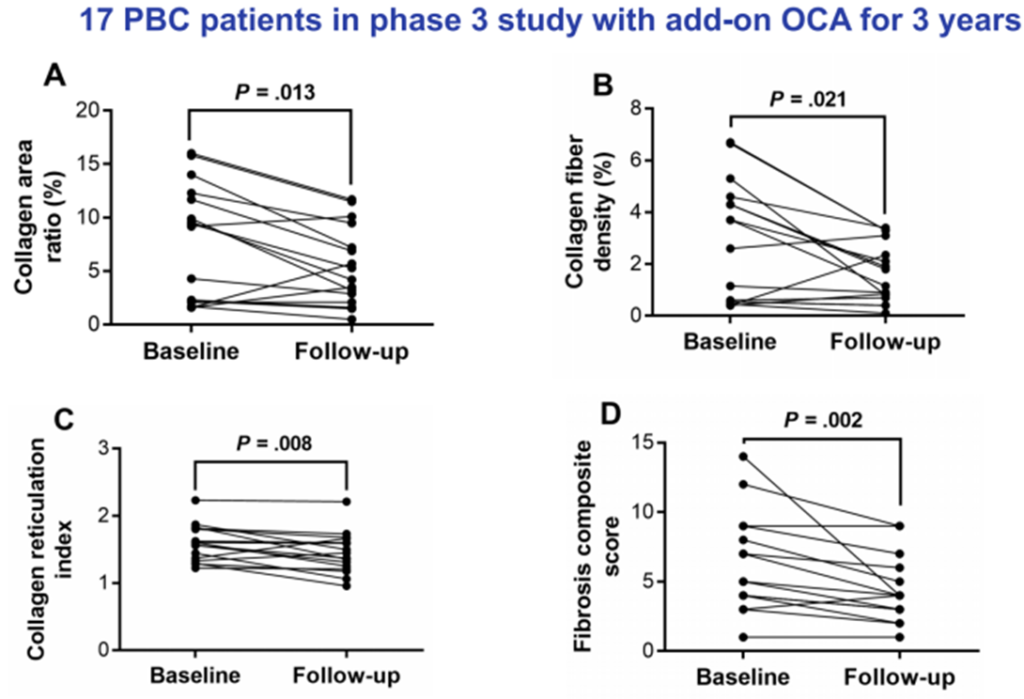
Figure 7. Three-Year Follow-Up Results for PBC Patients Receiving OCA (n=17)
(Sourced from the speaker’s presentation)
PPAR Agonists
Peroxisome Proliferator-Activated Receptors (PPARs) are part of the nuclear hormone receptor superfamily and are ligand-activated receptors, including three subtypes: PPARα, PPARγ, and PPARδ. PPARs are involved in the pathogenesis and progression of various liver diseases. Fibrates and other PPAR agonists can regulate bile acid synthesis through the activation of the PPAR pathway (Figure 8) [7].
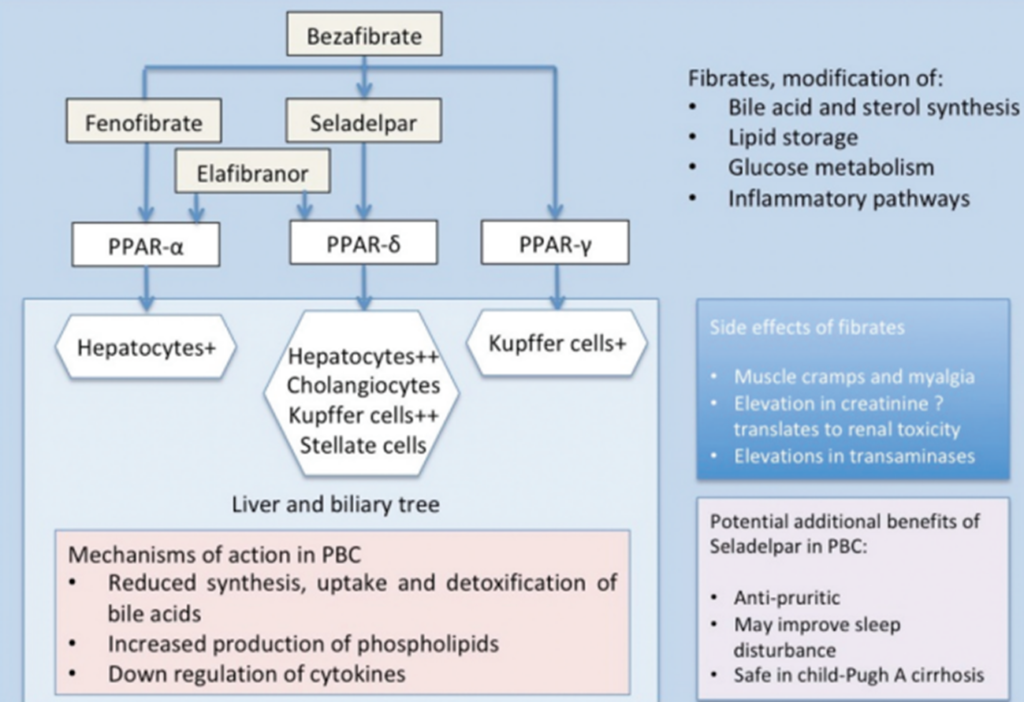
Figure 8. Mechanism of Action of PPAR Agonists
(Sourced from the speaker’s presentation)
Meta-analyses of randomized controlled trials and observational studies have confirmed that adding PPAR agonists is more effective than UDCA monotherapy in reducing ALP, GGT, IgM, and triglycerides [8-9], and they do not exhibit nephrotoxicity [10]. Randomized controlled clinical trials have also shown [11-12] that compared to the placebo group, the combination of bezafibrate and UDCA significantly lowers serum ALP levels in patients (Figure 9) and improves transplant-free survival in PBC patients (Figure 10). Additionally, new PPAR agonists for the treatment of PBC, including elafibranor (PPAR-ad), saroglitazar (PPARa/g), and seladelpar (PPARd), have demonstrated promising results in Phase II clinical trials.
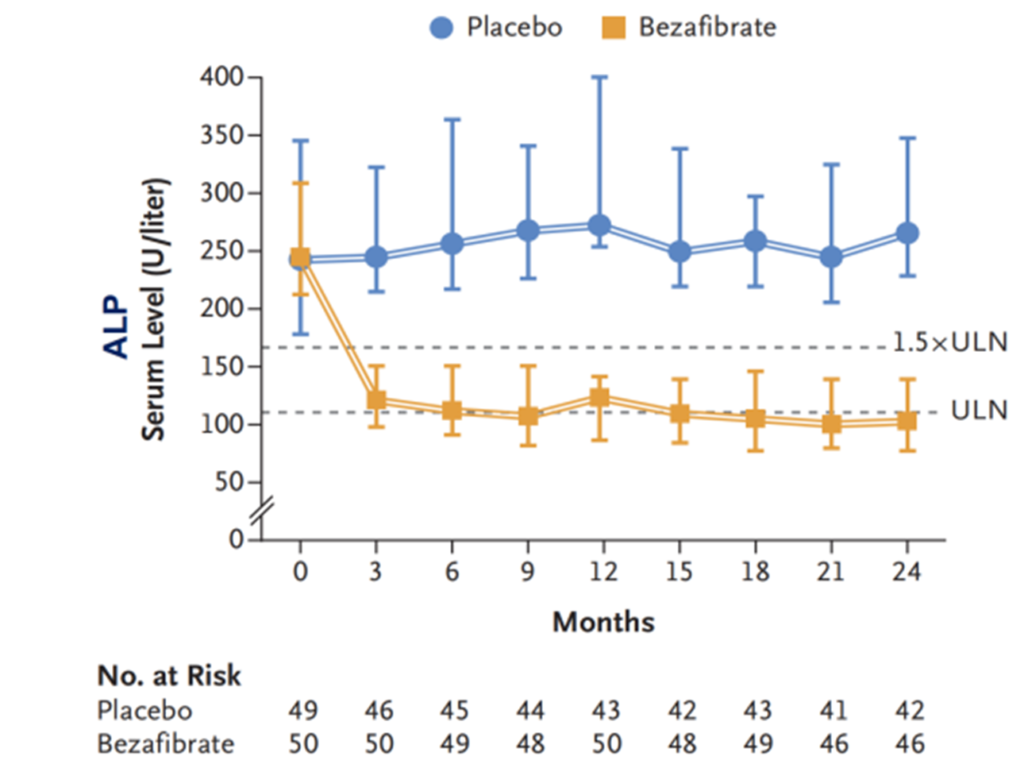
Figure 9. Impact of Bezafibrate Combined with UDCA on Patients’ ALP Levels
(Sourced from the speaker’s presentation)
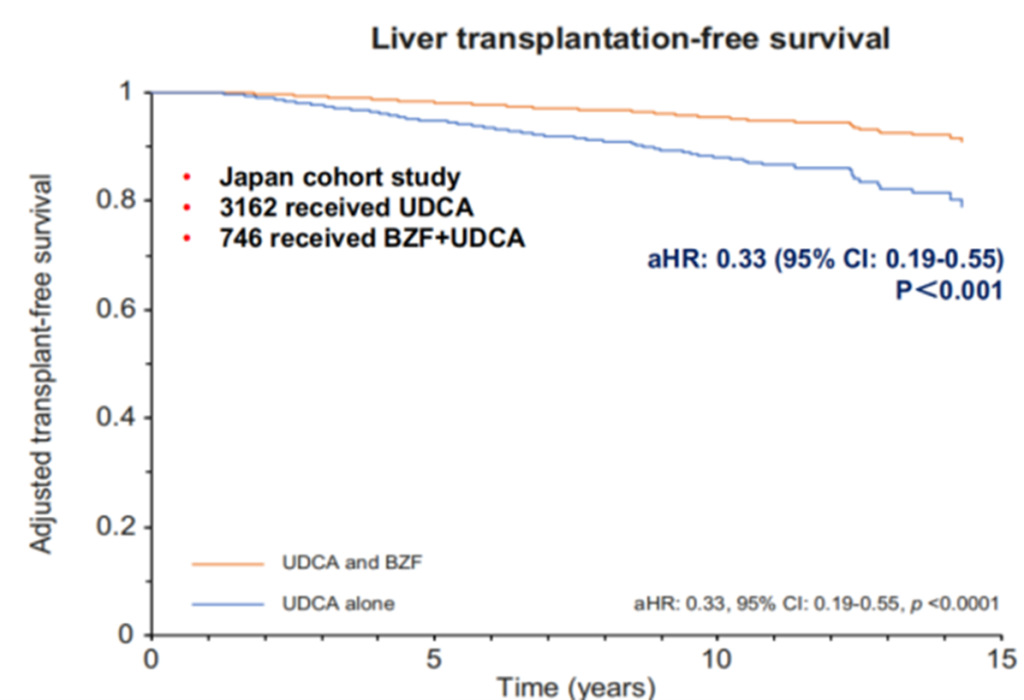
Figure 10. Impact of Bezafibrate Combined with UDCA on Patients’ ALP Levels
(Sourced from the speaker’s presentation)
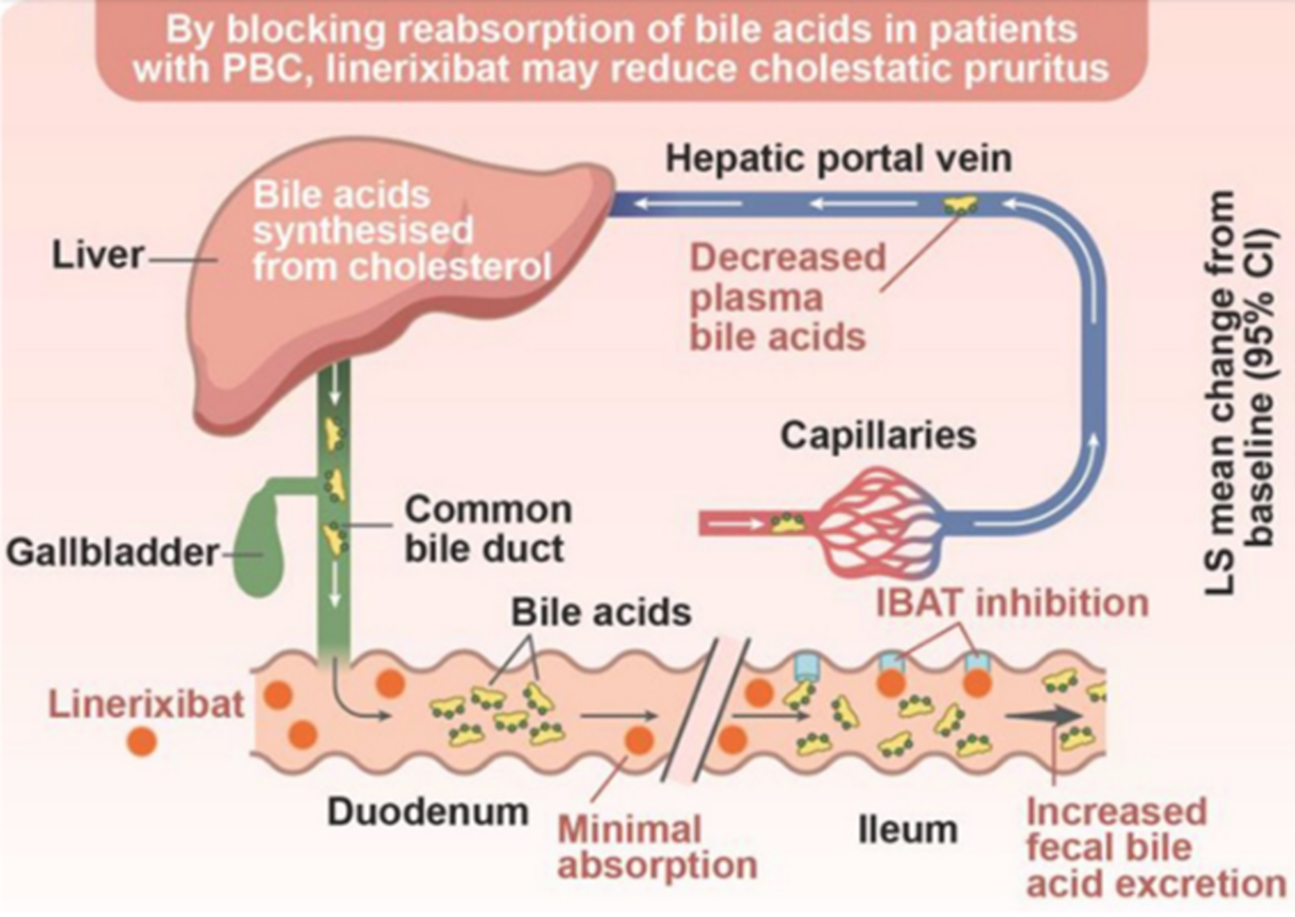
Figure 11. Mechanism of Action of IBAT Inhibitors
(Sourced from the speaker’s presentation)
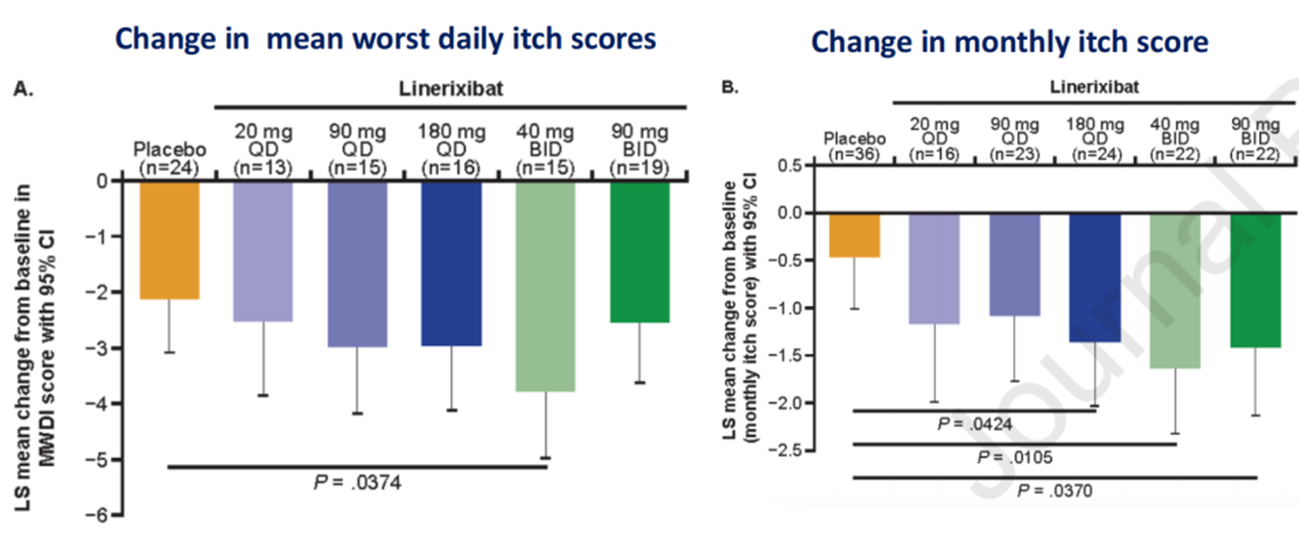
Figure 12. Impact of Linerixibat on Itching Symptoms in Patients
(Sourced from the speaker’s presentation)
Other Developments
Histological examination of liver tissues in patients with Primary Biliary Cholangitis (PBC) reveals infiltration by various immune cells, including T cells, B cells, macrophages, and natural killer cells. During the disease process, the intrahepatic small bile ducts are primarily targeted by immune cells, especially autoreactive T lymphocytes.
Double Negative T lymphocytes (DNT cells), also known as Double Negative T cells, are a subtype of T cells found in the peripheral blood of healthy individuals. They express the CD3 molecule but do not express CD4 and CD8 molecules. In recent years, due to their strong specific cytotoxicity against AML (Acute Myeloid Leukemia) tumor cells, DNT cells have emerged as a cellular therapy with significant anti-tumor potential. Recent studies have suggested [14-15] that DNT cells may play a potential role in the pathogenesis and treatment of PBC.
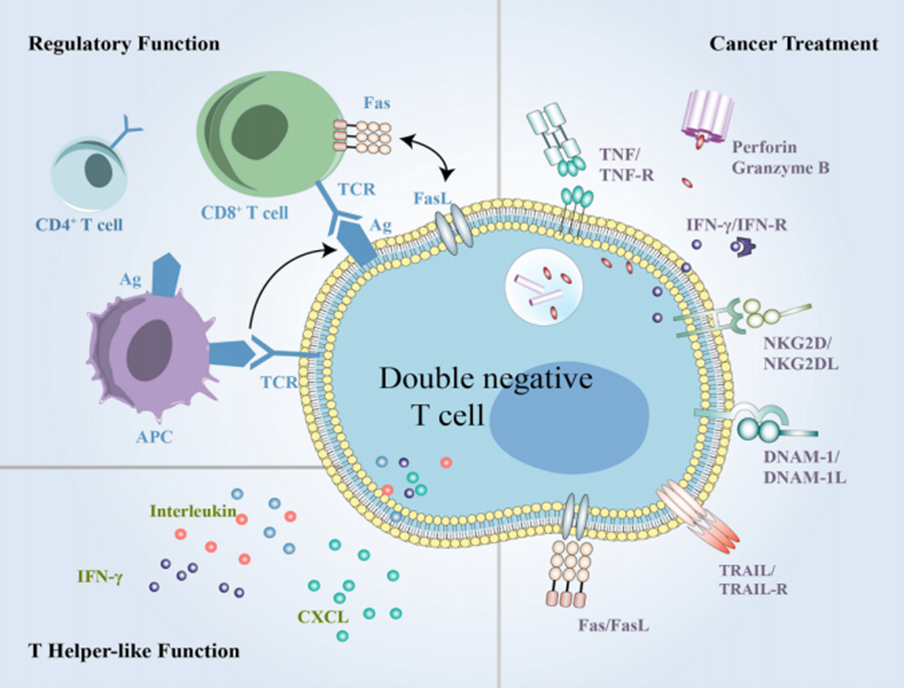
Figure 13. Mechanism of Action of DNT Cells
(Sourced from the speaker’s presentation)
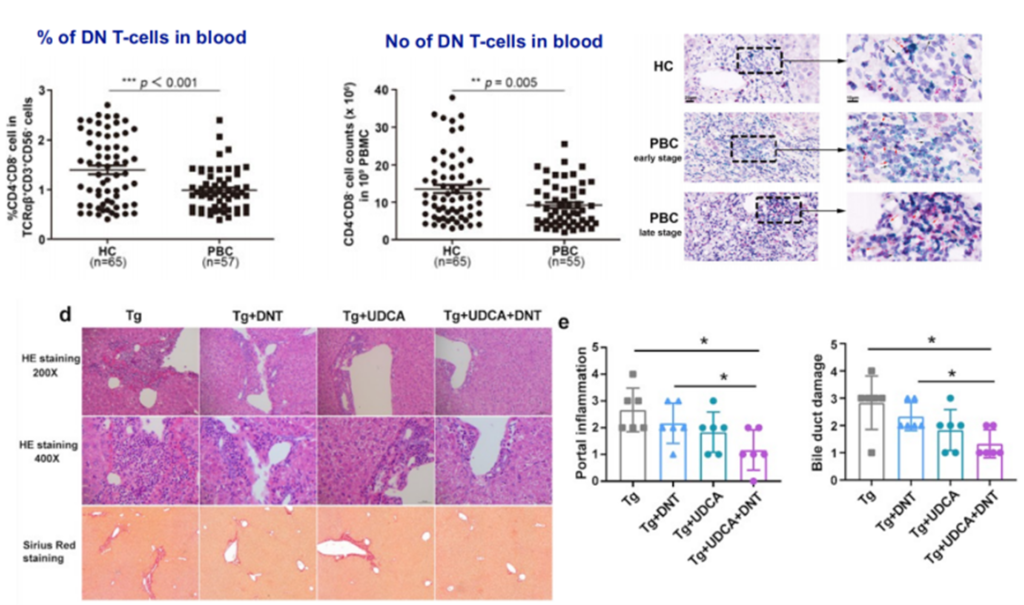
Figure 14. Role of DNT in the Treatment of PBC
(Sourced from the speaker’s presentation)
Summary
Currently, although the number of newly diagnosed cases of Primary Biliary Cholangitis (PBC) is increasing, there are relatively few effective drugs available for clinical treatment. As animal experiments and clinical studies continue to elucidate the role of bile acid metabolism, nuclear receptors, surface receptors, and transport proteins in the pathogenesis and progression of PBC, there is encouraging progress in the development of new drugs. It is expected that with the ongoing large-scale, randomized, controlled, prospective studies, the multifaceted treatment strategies targeting cholestasis will gradually be unveiled. The availability of a broader range of pharmaceutical options will better assist in the timely and effective control of clinical symptoms and delay the progression of the disease.
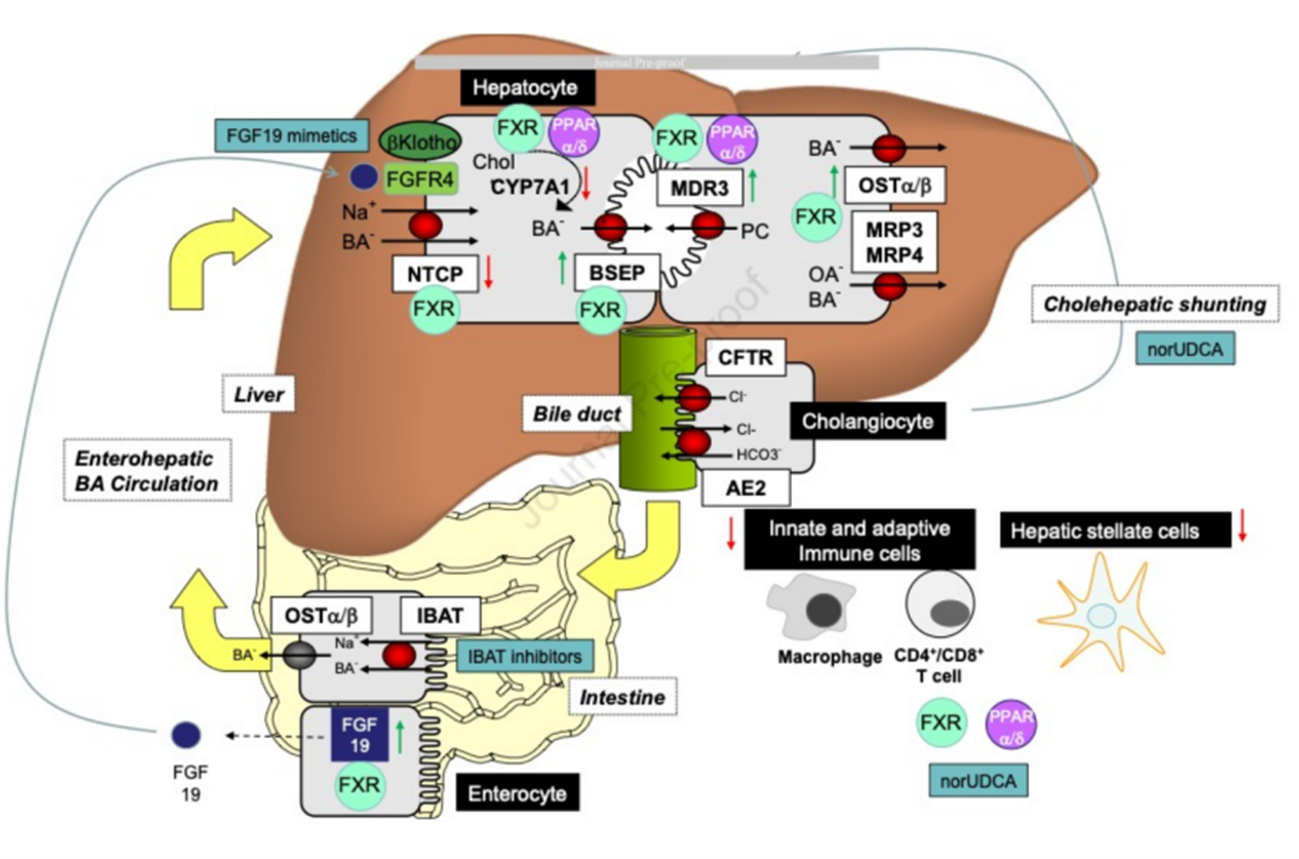
Figure 15. Summary of Potential Therapeutic Targets for PBC Treatment
(Sourced from the speaker’s presentation)
References :
- Lleo A, Wang GQ, et al. Primary biliary cholangitis[J]. Lancet, 2020, 396(10266): 1915-1926.
- Lv T, et al. J Gastroenterol Hepatol 2021.
- Zeng N, et al. Hepatol Int 2019.
- Lindor KD, et al. Hepatology. 2019;69:394-419.
- Harms MH, et al. J Hepatol. 2019;71:357-65.
- Nevens F, et al. N Engl J Med 2016;375:631-43.
- Wetten A, et al. Expert Opin Investig Drugs. 2022;31:1101-7.
- Grigorian AY, et al. Clin Res Hepatol Gastroenterol. 2015;39:296-306.
- Li C, et al. Ther Adv Chronic Dis. 2022.
- Yamaguchi M, et al. Hepatol Res. 2022, 52:522-531.
- Corpechot C, et al. N Engl J Med 2018;378:2171-81.
- Tanaka A, et al. J Hepatol. 2021;75:565-571.
- Levy C, et al. Clin Gastroenterol Hepatol. 2022.
- LI SX, et al. Liver Int, 2019,39:1755-1767.
- Zhang CP, et al. submitted MS.
TAG: APASL 2023, Master Class, PBC

